Finding Approximate Tandem Repeats in Genomic Sequences
Wexler, Y.1, Kashi, Y.2 and
Geiger, D.1
1 Department of Computer Science, Technion
2 Department of Biotechnology, Technion
Genomic sequences tend to contain consecutive copies of patterns
known as tandem repeats (TR). These occur due to DNA repair
systems and other mechanisms, not all fully understood.
Tandem repeats have proven useful as markers in genetic analysis due to
the high degree of polymorphism observed in their number (e.g. for DNA
fingerprinting applications [2]). Sometimes, such repeats are
known to be the cause of a disease like in the case of Fragile-X syndrome
and Huntington's disease.
A perfect tandem repeat is defined as a string
of nucleotides, which is repeated consecutively at least twice. Many
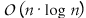 algorithms have been presented for finding perfect tandem
repeats. However, in practice, mutations, translocations, and other biological
events render the copies imperfect. This often results in approximate
tandem repeats (ATR) defined as a string of nucleotides repeated
consecutively at least twice such that, with sufficiently high probability,
they originated from the same string. Finding ATRs in a genome is clearly a
harder task than finding perfect repeats and has been addressed several times.
The IBM bioinformatics group, have presented the best results so far [3] by
altering their own TEIRESIAS pattern-finding algorithm.
algorithms have been presented for finding perfect tandem
repeats. However, in practice, mutations, translocations, and other biological
events render the copies imperfect. This often results in approximate
tandem repeats (ATR) defined as a string of nucleotides repeated
consecutively at least twice such that, with sufficiently high probability,
they originated from the same string. Finding ATRs in a genome is clearly a
harder task than finding perfect repeats and has been addressed several times.
The IBM bioinformatics group, have presented the best results so far [3] by
altering their own TEIRESIAS pattern-finding algorithm.
The algorithm we present herein has screening and verification
stages. In the screening stage, the possibility is evaluated of finding an
ATR of certain length, at a certain position, with sufficiently high
probability. In the verification stage, an alignment for each resulting
candidate is generated and subsequently accepted or rejected as an ATR
according to statistical criteria.
The screening stage uses a statistical model in which we
consider matching two adjacent
copies of a pattern of length t as a sequence of t independent
Bernoulli trials. We replace
the search for k contiguous successful nucleotide matches suggested by
Benson [1] with a less
stringent approach, as follows.
Consider a comparison to be a series of w
independent Bernoulli trials,
where w is a function of the pattern length t. Each trial
compares two aligned nucleotides
and the comparison passes if at least l(w) such trials succeed. Three
comparisons of length
w are performed for each starting offset i, where
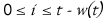 ,
to account for a single insertion, deletion or no-change at location i.
We define the success list
,
to account for a single insertion, deletion or no-change at location i.
We define the success list
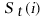 of i with respect to the
pattern length t
to be the set of integers
of i with respect to the
pattern length t
to be the set of integers
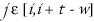 for which the
comparison starting at
position j succeeded. The cardinality of
is called the score of i with respect to t.
for which the
comparison starting at
position j succeeded. The cardinality of
is called the score of i with respect to t.
In order for a position to pass the screening and
become a candidate for
verification, it has to satisfy several statistical criteria regarding the
P-value of its score and
a valid distribution of successful comparisons. We perform these
comparisons on an entire
genome of size n for all pattern lengths up to some
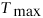 in time
in time
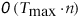 . This complexity is achieved because, instead of computing each comparison
separately, they are computed incrementally while sliding over the genome.
. This complexity is achieved because, instead of computing each comparison
separately, they are computed incrementally while sliding over the genome.
The verification stage, when given a candidate
ATR starting at position
i with pattern length t, generates an alignment between the
pattern starting at position
i and the one starting at position i+t, both of length t,
using a predetermined
scoring system. If the alignment score is over a threshold, which depends on
the scoring system, then
the candidate is accepted as an ATR.
Results
The algorithm presented herein, aside
of having a linear time
complexity in genome size n when fixing
, also performs
well on real examples. When running over the yeast chromosome 1, which is the
same genomic sequence used
by IBM bioinformatics group to establish their results, using the same scoring
system for verification as
they did, and finding ATRs with length ranging from 10 to 300, the following
facts emerge:
-
Our algorithm found more than twice as many ATRs as was previously
reported, finding also all the
ones known before.
-
A low rate of false positive candidates occurs during the screening stage
(~3%). Consequently, the
running time was less than 1% of the time reported by the IBM group, on a
machine with no greater power.
-
Our algorithm is parameter-free and does not require additional input from
the user, aside of
biological data, sequences, and a desired scoring system for alignment.
References
-
Benson, G. 1998. Tandem repeats finder: a program to analyze
DNA sequences. Nucleic Acid Research, vol. 27 pp. 573-580.
-
Inman, K. and Rudin, N. 1997. An introduction to forensic DNA analysis.
{\em CRC press,} Boca Raton, Florida.
-
Stolovitzky, G., Gao, Y., Floratos, A. and Rigoutsos, I. 1999. Tandem
repeat detection using pattern discovery, with applications to identification
of yeast satellites. {\em IBM T.J.Watson Research Center.}
e-mail:
ywex@cs.technion.ac.il
e-mail:
kashi@tx.technion.ac.il
e-mail:
dang@cs.technion.ac.il