Profile Hidden Markov Model Analysis
[ Program Manual | User's Guide | Data Files | Databases ]
Profile analysis has long been a useful tool in finding and aligning distantly related sequences and in identifying known sequence domains in new sequences. Basically, a profile is a description of the consensus of a multiple sequence alignment. It uses a position-specific scoring system to capture information about the degree of conservation at various positions in the multiple alignment. This makes it a much more sensitive and specific method for database searching than pairwise methods, such as those used by BLAST or FastA, that use position-independent scoring.
Hidden Markov modeling, a technique that has been used for years in speech recognition, is now being applied to many types of problems in molecular sequence analysis. In particular, this technique can produce profiles that are an improvement over traditionally constructed profiles.
Profile hidden Markov models (HMMs) have several advantages over standard profiles. Profile HMMs have a formal probabilistic basis and have a consistant theory behind gap and insertion scores, in contrast to standard profile methods which use heuristic methods. HMMs apply a statistical method to estimate the true frequency of a residue at a given position in the alignment from its observed frequency while standard profiles use the observed frequency itself to assign the score for that residue. This means that a profile HMM derived from only 10 to 20 aligned sequences can be of equivalent quality to a standard profile created from 40 to 50 aligned sequences. In general, producing good profile HMMs requires less skill and manual intervention than producing good standard profiles.
The HMMER (pronounced hammer) package developed by Sean Eddy of Washington University in St. Louis, Missouri, is a set of programs that allow you to create and manipulate profile HMMs and databases of profile HMMs (HmmerBuild, HmmerConvert), perform sensitive searches of sequence and profile HMM databases, (HmmerSearch and HmmerPfam) and create multiple sequence alignments efficiently (HmmerAlign). In collaboration with Dr. Eddy, GCG has incorporated these programs into the Wisconsin Package.
A profile HMM is a
linear state machine consisting of
a series of nodes, each
of which corresponds
roughly to a position (column)
in the alignment from which
it was built. If
we ignore gaps, the
correspondence is exact -- the
profile HMM has a node
for each column in the
alignment, and each
node can exist in one
state, a match state.
(The word "match" here implies
that there is a position
in
the model for every position
in the sequence to be
aligned to the model.)

A profile HMM has several types of probabilities associated with it. One type is the transition probability -- the probability of transitioning from one state to another. In a simple ungapped model, the probability of a transition from one match state to the next match state is 1.0 and the path through the model is strictly linear, moving from the match state of node n to the match state of node n+1.
There are also emissions probabilities associated with each match state, based on the probability of a given residue existing at that position in the alignment. For example, for a fairly well-conserved column in a protein alignment, the emissions probability for the most common amino acid may be 0.81, while for each of the other 19 amino acids it may be 0.01. If you follow a path through the model to generate a sequence consistent with the model, the probability of any sequence that is generated depends on the transition and emissions probabilities at each node.
In order to model real sequences, we also need to consider the possibility that gaps might occur when a model is aligned to a sequence. Two types of gaps may arise. The first type occurs when the sequence contains a region that is not present in the model (an insertion in the sequence). The second type occurs when there is a region in the model that is not present in the sequence (a deletion in the sequence). To handle these cases, each node in the profile HMM must now have three states: the match state, an insert state, and a delete state. The model also needs more types of transition probabilities: match->match, match->insert, match->delete, insert->match, etc.
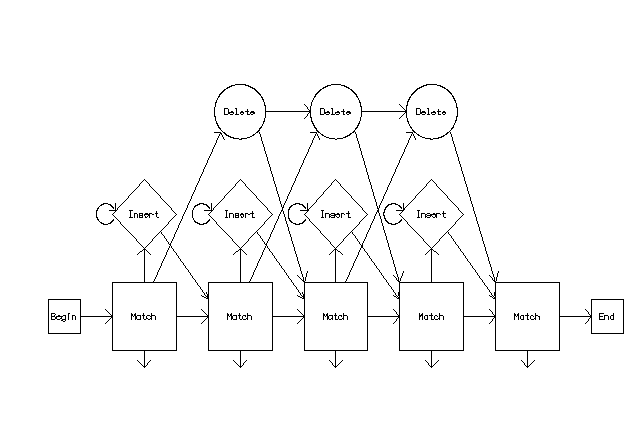
Aligning a sequence to a profile HMM is done by a dynamic programming algorithm that finds the most probable path that the sequence may take through the model, using the transition and emissions probabilities to score each possible path.
In general, if the sequence is equivalent to the consensus of the original alignment, the path through the model will pass from match state to match state in a linear fashion. If the sequence contains a deletion relative to the consensus, the path passes through one or more delete states before transitioning to the next match state; if the sequence contains an insertion relative to the consensus, the path passes through an insert state between two match states.
For example, if a sequence contains an insert that occurs between nodes 5 and 6 of the model, the path transitions from the node 5 match state to an insert state. It remains in the insert state and "consumes" residues in the sequence until it reaches the residue in the sequence that corresponds to node 6 in the model. At this point the path transitions from the insert state to the node 6 match state.
Similarly, if the sequence contains a deletion so that it has no residues corresponding to nodes 12 through 15 of the model, the path transitions from the node 11 match state into a delete state, then transitions through additional delete states until it can transition to the match state of node 16 of the model.
Profile HMMs can be aligned to a sequence either globally (the whole profile HMM aligns to the sequence) or locally (only part of the profile HMM need be aligned with the sequence). The alignment type is actually part of the model, so you must specify whether the model is to be global or local at the time the model is built, not at the time the model is used. (See HmmerBuild documentation for more details.)
Because a profile HMM can serve as a representation of a sequence family or sequence domain, the most common application is to compare profile HMMs and sequences. These types of comparisons are more likely to identify distant homologs than sequence vs. sequence comparisons used in most database search programs.
For example, you can use HmmerPfam to compare your sequence to a database of profile HMMs representing known sequence families and known sequence domains. A match to one of these profile HMMs can help you identify your sequence and determine its function. The curated Pfam ("Protein families") database contains a large number of global profile HMMs representing known protein families, while the PfamFrag database contains local profile HMMs for these same families.
Similarly, you can create a profile HMM representing a domain or sequence family in which you are interested, then use this profile HMM as a query to search a sequence database with HmmerSearch to see if any other sequences possess this domain.
Another use for profile HMMs is to create a multiple alignment of a large number of sequences more quickly than by using standard methods. HmmerAlign uses a small seed alignment of representative sequences to create a profile HMM which is then used as a template for aligning the full set of sequences.
There are nine programs in the GCG adaptation of the HMMER package. The following five are used to create and manipulate profile HMMs:
HmmerBuild -- creates a profile HMM from a set of pre-aligned sequences. The profile HMM can be appended to a file containing other profile HMMs in order to create an HMM database file.
HmmerCalibrate -- calibrates an existing profile HMM or profile HMM database so that searches performed with it will be more sensitive.
HmmerConvert -- converts a profile HMM created by HmmerBuild into other formats.
HmmerIndex -- indexes a profile HMM database so that profile HMMs can be retrieved from it easily with HmmerFetch.
HmmerFetch -- extracts a profile HMM from an indexed profile HMM database into a file.
The remaining four programs are used for analyzing data:
HmmerSearch -- searches a sequence database with a profile HMM query.
HmmerPfam -- searches a profile HMM database with a sequence query. The profile HMM database file may be one you created as well as the Pfam database created by Eddy and collaborators.
HmmerAlign -- efficiently creates a large multiple alignment from a small seed alignment and a collection of unaligned sequences.
HmmerEmit -- randomly generates sequences that match a given profile HMM.
Pfam - A database of protein domain family alignments and HMMs Copyright (C) 1996-2000 The Pfam Consortium.
1. Eddy, S.R., et al. (1996). Hidden Markov Models. Current Opinion in Structural Biology,
6; 361-365.
2. Durbin, R., Eddy, S., Krogh, A., and Mitchison, G. (1998). Biological Sequence Analysis.
Probabilistic Models of Proteins and Nucleic Acids, Cambridge University Press, Cambridge, UK.
3. Eddy, S.R. (1998). Profile hidden Markov models. Bioinformatics, 14; 755-763.
[ Program Manual | User's Guide | Data Files | Databases ]
Technical Support: support-us@accelrys.com
or support-eu@accelrys.com
Copyright (c) 1982-2002 Accelrys Inc. A subsidiary of Pharmacopeia, Inc. All rights reserved.
Licenses and Trademarks Wisconsin Package is a trademark and GCG and the GCG logo are registered trademarks of Accelrys Inc.
All other product names mentioned in this documentation may be trademarks, and if so, are trademarks or registered trademarks of their respective holders and are used in this documentation for identification purposes only.
![]()